
ISSN (0970-2083)
ISSN (0970-2083)
A. Popov1, A. Obkhodsky2* and S. Kuznetsov2
1LLC “Depos”, 634033, Tomsk, Russian Federation
2National Research Tomsk Polytechnic University, 634050, Tomsk, Russian Federation
Received date: July 06, 2016; Accepted date: August 19, 2016
Visit for more related articles at Journal of Industrial Pollution Control
The paper presents mathematical models on the basis of first-principles calculation procedures (ab initio) for the description of the atomic structure properties of materials. The general calculation procedure of materials’ properties with application of the Hartree method and Density Functional Theory was developed on the basis of the carried out analysis. The procedure was applied in the form of the algorithm and introduced in “Antares” – a program package for material modeling which functions on the basis of GRID network of distributed computing. Experimental verification of the developed procedure was carried out on the example of Al3Ti system design by LCAO-CO method with the use of STO-3G functional. Al3Ti grid parameters were defined in the course of experimental validation. Estimated value of grid a parameter was 4,889 Å.
Ab initio, The hartree-fock-roothaan method, Density functional theory, Crystal calculation, LCAO-MO; LCAO-CO, Parameters calculation procedure, Basis sets.
It has appeared more opportunities for complicated technical and scientific calculations with the growth of IT equipment efficiency. At the same time, the theoretical foundation is being expanded to increase the efficiency of already existed calculation assessment techniques of atomic structure properties and to create new ones. There is nearly always demand for new materials on the market, which have unique mechanical and strength properties.
The development of a durable, light, heat-resistant and cost-efficient material is among tasks aimed on finding new materials, which can be used in deferent branches of industrial production, for example, in aircraft industry or in spacecraft production. There are also tasks for creation of heat-resistant materials for nuclear industry.
The obtaining of basic theoretical knowledge about the properties of one or another material can release scientists from carrying out costly experimental researches. Computer modeling is considered to be efficient at estimation of materials properties in conditions the reproduction of which is hard or impossible in an experiment.
The use of specialized systems of distributed computing allows to reduce considerably execution time of calculation. The distributed computing functions of the existing program packages for quantum-chemical calculations are provided by means of additional software which functions on separate and unique software platforms of supercomputers. The program package “Gaussian” is a popular software product which supports a large number of calculation methods. It provides high precision of calculated results, it is cross-platform and it operates input and output data with unified format which is supported by other auxiliary programs.
The aim of this paper was to create a generalized method for property calculation of atomic structures of solid materials. The area of application of this procedure is national program packages for quantum-chemical modeling including “Antares” package which is being created. The experimental approbation of the developed procedure was carried out on the example of the properties calculation of Al3Ti system (Grishaeva et al., 2014).
The Calculation Procedure of Material
Today there are a lot of methods to calculate atomic structures. Every method has its specialization, precision, validity criterion and etc. The most known ones among non-empirical are the Hartree-Fock (HF) method and Density Functional Theory (DFT). The HF method preceded the invention of DFT and it was quite self-sufficient in the determination of properties and precision. But it was efficient only for elementary compounds and elements. The complexity of mathematical apparatus was growing exponentially during the procedure of medium systems calculations, consequently, the requirements to the IT equipment had grown. Moreover, the precision of calculations had dropped significantly. The loss of precision in the HF method was due to the fact that it does not allow for electron correlation. There are modified HF methods which allow for electron correlation, however, in the comparison with DFT method the HF ones are of inferior speed in calculations. The reason is that the HF method describes a system by means of many-electron wavefuction while DFT method describes it with the use of electron density. It should be noted that DFT method is not entirely analytically reliable what is proved in the paper (Sarry and Sarry, 2012) devoted to W. Kohn’s statements.
The sequence scheme of materials’ property calculations can be generalized as shown in Fig. 1. The first step is to choose components. The initial condition of a system is determined on the second step, for example, temperature (which then might low to 0). Oscillatory processes make a less contribution in comparison with Coulomb interactions while crystal grid is being formed but it is necessary to consider this factor in order to increase calculation precision.

Fig. 1: The generalized scheme of materials’ property calculations in modeling procedure.
The calculation time highly depends on the next step which is the choice of a basis set, because the calculations would take a lot of time during the choice of initial data which significantly differ from the solution of an optimization task. On the whole, this stage determines the choice of advance estimation of crystal grid and atomic orbitals.
The next step is to solve the Schrödinger equation. As it is impossible to solve the equation for systems with more than one electron in an atom, so approximations were suggested, which do not consider some properties of electrons but at the same time the solution of the Schrödinger equation in some variants becomes possible.
Two methods of first principles calculations can be used to estimate atomic structures: the Hartree approximation is used in the first one and Density Functional Theory is used in the second. The vital difference between those methods is that in the Hartree method every electron is considered to be isolated one and it is influenced by a field which consists of collection of fields created by a nuclei. At the same time correlation effects that can be determined after calculations are not considered. Also, in some cases, taking into the consideration these effects the calculations can be made again, this will increase precision but the calculation procedure will take more time.
DFT is a method which demands less computing resources. In this method collection of electrons is considered as a functional the form of which depends on the given task. Despite the advantage in calculation time, DFT method is inferior to the Hartree method in precision in some calculations. Recently, DFT precision is commensurable to precision of the Hartree method because the theoretical foundation exactly for DFT method is being developed more successfully that allows to optimize calculations and take into the account a larger number of quantum effects. It should be noted that this method allows for correlation effects before the calculations and this is the one of the most successful solutions how to decrease calculation time and to increase precision. In other words, if these effects are not considered in the Hartree method after calculations, so DFT method will exceed it among all parameters. The differences between these two methods are thoroughly discussed in Klekovkina V.V. paper (Klekovkina and Aminova, 2009).
The results are verified after calculations. At first geometric entities are checked. To what extent the data of previous integration differs from the data that had been got for the optimization moment of an atomic system. If differences are too large, it is worthwhile to repeat calculations with newly received data about a crystal grid.
As a rule, for the final verification a difference of received quantity from experimental quantities is analyzed. If both previous stages were successful, the results can be use for determination of different material parameters. For example, DFT method is used to calculate geometrical parameters of a crystal grid (Babkin et al., 2011), and also to calculate energy connection between its points (Iljasov and Fam, 2014; Nelasov and Lipninskij 2011; Kravcova et al., 2011; Kazakova, 2009). According to the results of parameter calculations of atomic structures it can be calculated strength (Iskandarov and Umeno, 2012; Zavodinskij, 2010), electron structure (Kravcova et al., 2011; Starodubceva et al., 2012; Harchenko et al., 2013; Rakitin et al., 2010), band structure and magnetic characteristics (Babkin et al., 2011), formation of grain boundary and its properties (Verhovyh and Mirzoev, 2013), optical properties (Gazhulina and Marychev, 2012), surface energy (Mamonov et al., 2011) and others.
Further in this paper the calculation sequence of Al3Ti atomic structure will be carried out with the use of the modified Hartree-Fock method. So far as DFT is similar in many points to the HF method a program code can be modified for DFT.
Automation of Calculation with the use of Hartreefock Method
The Hartree-fock method is one of the methods which allows to get a solution for a system consisting of many particles. As we have already mentioned, the idea of the Hartree method is that every separately taken electron moves independently from others but at the same time it is influenced by external field of nuclei and other electrons. Electrostatic field produced by average charge density is a replacement action of this electron for other electrons. The Hartree method was modified by Fock and Slater as a result the Pauli principle was considered in the creation procedure of many-particle wavefunction that is why the Hartree-Fock method allows for exchange interaction.
If each electron has its own wave function (orbital) ÃÂÃâ¢i, the complete wavefunction of all N electrons can be presented in the form of the Slater determinant (Minkin et al., 1997):
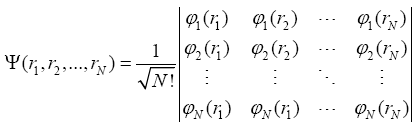
In the case of substitution of this many-electron wavefunction in the Schrödinger equation and applying the Ritz variation principle we get:
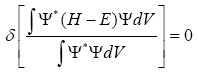
If we consider the orthonormality of wavefunctions ÃÂÃâ¢i, we will get the Hartree-Fock equation which is also an expression for Fock operator:
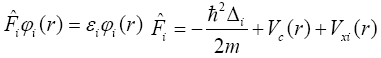
Vc(r) is the Coulomb’s potential energy in the point r,
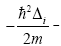 operator of kinetic energy,
operator of kinetic energy, 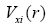 - exchange potential which has the product of all one-electron wavefunctions:
- exchange potential which has the product of all one-electron wavefunctions:
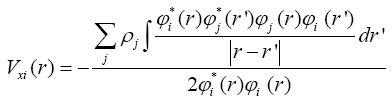
The dependence of  operator on one-electron wave functions considerably obstructs the solution of the Hartree-Fock equation. Such kinds of equations are solved in an iterative way: are found with the use of initial functions ÃÂÃâ¢i, then the set of one-electron wave functions are found. These stages are repeated until convergence criterion (self-consistency) is achieved (Abarenkov et al., 1989).
operator on one-electron wave functions considerably obstructs the solution of the Hartree-Fock equation. Such kinds of equations are solved in an iterative way: are found with the use of initial functions ÃÂÃâ¢i, then the set of one-electron wave functions are found. These stages are repeated until convergence criterion (self-consistency) is achieved (Abarenkov et al., 1989).
For a long time the Hartree-Fock method has been the best way to calculate atomic structures, however, problems appear problem in the case of huge systems due to a large number of crossed exchange integrals. However, this method allows for exchange interaction, it does not allow for correlation effects what is considered to be the main disadvantage of this method.
The Roothaan method has become the most successful method for solution of the Hartree-Fock equations (Baranovskij, 2008). Gausssian basic functions are used in the Roothaan method as they are used in Density Functional Theory method.
The Choice of the Basic Set
The basic set is a set of functions with the help of which atomic orbitals are described. Atomic orbitals are one-electron wavefunctions which are determined according to the solution of the Schrödinger equation. Any atomic orbital defines “spatial” electron density, and the basic set defines molecular orbitals and consequently electron density in the given space.
The STO-nG basic set is the simplest one. In a fundamental way it is an atomic orbital of Slater kind, which is approximated with n functions of Gaussian type. In other word, any atomic orbital consists of a sum of n Gaussian functions, at the same time the coefficients of Gaussian functions are selected to approximately describe the behavour of Slater orbitals with the help of linear combinations.
STO-3G is the most common among family of STO-nG basis set because testing calculations show that the results practically coincide with n>3 and necessary precision is not achieved with n = 2.
In a minimal basis set it is impossible to change orbital size depending on molecule size and this is a disadvantage of such kind of sets that lead to mistakes in approximate calculations of ions and neutral molecules. These disadvantages are eliminated by means of atomic orbitals increasing with the help of bi-exponential (doublezeta) or split valence basis sets. Atomic orbitals consist of two parts in biexponential basis sets: external (more diffusive) and internal (more compact). Valence orbitals are divided only in split valence basis sets. Basis set 6-31G is the most common among the last ones. An orbital of a core consists of six Gaussian functions, valence orbitals are divided into a compact function consisting of tree Gaussian functions and a diffusive function consisting of one Gaussian function.
Two basis sets are often used simultaneously – the geometry is optimized with the help of one, the calculation for one geometric configuration is carried out with the help of another. For example, in 6- basis 31G//STO-3G the first function is performed by STO-3G basis and the second one is performed by 6-31G. Suck kind of differentiation make it possible to increase calculation precision.
There exist modifications of above mentioned bases and there are also hundreds of others. We should note that the basis choice is determined by the necessity of calculation precision and also by computing resources of IT equipment. It is necessary to remember that the consumption of computer time is proportional to the number of basis functions that is raised to the fourth power. As a rule, geometry optimization is performed on simple bases, and then by means of more complicated ones, it is also carried out the calculation of corrections which are connected with electron correlation. The calculation time decreases with the use of this method and in most cases precision is comparable with calculations in wider basis with full optimization.
Quite good results at optimization of elements of the third period are achieved due to relatively simple basis 3-21G*. In advance, it is recommended to optimize the geometry by means of molecular dynamic method in order to optimize big molecules if there is no opportunity to use more complicated basis.
Basis STO-3G is used to calculate Al3Ti material because of its simple application and because it is easily modified into any other basis set.
The Hartree-Fock method was applied to find oneelectron functions of molecular orbitals. Numerical value lists were the result because there is no solution of the equation in an analytical form. One of the efficient approximations is LCAO-MO approximation (molecular orbitals as linear combinations of atomic orbitals). According to LCAO-MO every molecular orbital is described as a linear combination of atomic orbitals forming a molecule that is why LCAO-MO is the simplest method to determine molecular orbitals.
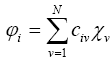
LCAO-MO approximation is applied in the Hartree- Fock-Roothaan method, which in turn can be of two types: for open and closed electronic shells.
In that case the Roothaan equation would have a look in the following way:
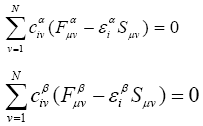
Matrix elements 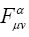 and
and 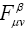 will be written in the following way:
will be written in the following way:
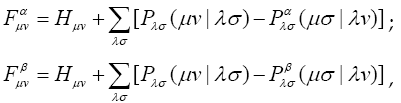
Whereas
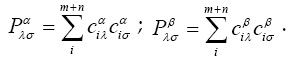
Basis STO-3G was chosen as a basis set and it describes the elements of the chosen system quite precisely. In that basis Slater-type orbitals are described as a combination of three Gaussian functions:
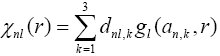
Approximation LCAO-CO (crystal orbital as a linear combination of atomic orbitals) is introduced by analogy with LCAO-MO – analogue of the Hartree- Fock-Roothaan method for crystals. On the basis of the method results we can link properties of atoms forming a crystal to the crystal itself.
In LCAO-CO one-electron functions for the crystal are constructed as linear combinations of atomic orbitals sums in blocks:
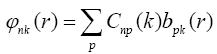
Coefficients 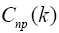 are found with the help of variational principle from the Hartree-Fock- Roothaan equations:
are found with the help of variational principle from the Hartree-Fock- Roothaan equations:
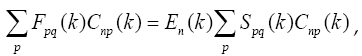
and besides it comes out as N systems for crystals instead of such a system for a molecule.
Matrix 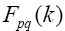 is found as a sum of matrixes
is found as a sum of matrixes 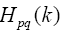
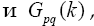 the elements of which are determined in the following way:
the elements of which are determined in the following way:
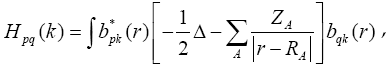
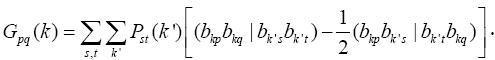
Lattice sums of matrix elements calculated with functions from the zero (central) cell:
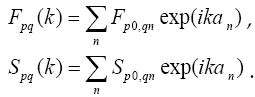
While solving the Hartree-Fock-Roothaan equations for crystals it is necessary to know 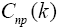 of the whole reduced Brillouin zone for fixed value because the specified form of matrix elements as follows:
of the whole reduced Brillouin zone for fixed value because the specified form of matrix elements as follows:
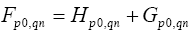
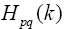 are Hamiltonian elements for the crystal which contains energy of electron interaction with cores V(r), and also it contains kinetic energy. Twoelectron component
are Hamiltonian elements for the crystal which contains energy of electron interaction with cores V(r), and also it contains kinetic energy. Twoelectron component 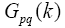 contains summation according to filled state:
contains summation according to filled state:
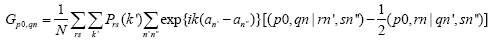
As a result, for self-consistent calculation of the electron crystal structure it is necessary to summarize a large number of occupied electronic states on every stage of integration process and that is why this calculation is more complicated than the calculation of molecules.
The model described by the given equations was applied in program package Antares for atomic system Al3Ti. It was carried out the calculation of geometrical parameter a of the crystal grid; in the result we got the value which is equal to 4,889 Å, what is 1,053 Å more than the 3,836 Å experimental value (Nikolskij, 1966). According to the result we can make a conclusion that the Hartree-Fock method for crystals which is used jointly with STO- 3G basis calculates geometrical parameters of the chosen atomic system Al3Ti with relative error 25% because of error accumulation during the system self-consistency procedure.
The work is performed with financial support of the Ministry of Education and Science of the Russian Federation. Grant agreement No. RFMEFI57814X0095 as of November 28, 2014.
Copyright © 2026 Research and Reviews, All Rights Reserved