
ISSN (0970-2083)
ISSN (0970-2083)
Thirunavukkarasu M1*, Dinakaran K2, Sathishkumar E.N3 and Gnanendra S4
1Department of Computer Science, Mahendra Arts & Science College (Autonomous), Kalippatti, Research and Development Center, Bharathiar University, Coimbatore, Tamil Nadu, India
2Department of Computer Science and Engineering, PMR Engineering College, Adayalampattu, Chennai, Tamil Nadu, India
3Department of Computer science, Periyar University, Salem-11, Tamil Nadu, India
4Department of Biotechnology, Yeungnam University, Gyeongsan, Gyeongbuk, 38541, South Korea
Received Date: 17 June, 2017 Accepted Date: 22 August, 2017
Visit for more related articles at Journal of Industrial Pollution Control
Machine learning algorithms are significant computational methods that are used to extract the knowledge from data. In general, neural networks and support vector machines (SVM) are the generally adopted techniques in the knowledge prediction of biological data. The availability of complete bacterial genomes information and the complexity in determining the virulence factors raised the urgency in the need of computational tools to predict the virulence factors. Thus in this study, the predictive capability of SVM and Back propagation network (BPN) algorithms and their reliability were determined by a widely used cross-validation tests in statistics. While a comparative study on the performance of the methods based on the feature representation are analyzed along with these classiïìÃÂcation methods. SVM classifiers was trained and optimized with different kernel parameters and sequence features like composition of amino acid, combination of amino acids forming dipeptides and composite methods. In addition, BPN classifiers were also trained for the same dataset. A ten-fold cross-validation was used to evaluate the performance of both SVM and BPN classifiers. The effect of feature representation methods (AAC, DPC and Composite) on the classiïìÃÂcation performances of SVM and BPN were evaluated. The SVM classiïìÃÂers trained with AAC features revealed that the accuracy of 79.13 %, while it is of 86.56% for BPN. The prediction accuracy of SVM is almost 10% and 3% greater than the BPN while using DPC and composite features respectively. Whereas, the specificity and sensitivity of SVM were found to be low than that of BPN. Thus suggesting the usages of BPN over SVM classifiers as the best classifier for predicting the proteins sequence based on their compositions.
Machine learning algorithms, SVM, BPN, Protein sequence, virulent
The recent advances in computational methods and algorithms have put forth the implication of machine learning techniques as all purpose methods to extract the knowledge from data. The machine learning techniques such as neural networks and support vector machines (SVM) are the most widely adopted methods by computational biologists to extract the knowledge from the available biological data. In this category, an artificial neural network that can simulate the structure and functional aspects of biological data as neural networks has gained more attentions (Bishop, 2006). This mathematic model can change its structure based on the information that flows through the network from external and internal while the system is trained. These methods are usually employed to establish the relationships between inputs and outputs or to find complex patterns in data (Hertz, et al., 1991). The availability and dissemination of biological data has opened challenges to many virulence researches. In recent years, the continuous usage of antibiotics and disinfectants in nosocomial infections resulted in the dramatic increase in the emergence of multidrug resistance of the bacteria. In almost all of the pathogenic organisms, Virulence Factors (VFs) play a major role in exhibiting its pathogenic ability. VFs are the kind of molecules that are associated with pathogenic microorganisms to accelerate the disease causing ability of an organism (Wu, et al., 2008). Traditionally, several biochemical tests are used to identify the bacterial pathogens and their VFs before the effective prophylaxis is carried out. However, these laboratory science methods make the identiïìÃÂcation and veriïìÃÂcation of virulence factors are often costly and time taking process. Thus the need and development for the automatic algorithm as a most robust and consistent in predicting the virulence factors are in demand and need.
Though many tools that can classify the protein sequences as virulent factors and non-virulent factors are available, the prediction ability of these tools in terms of sensitivity, specificity and accuracy remain as a challenging factor (Lin, et al., 2009). In line with this, many research groups have documented their findings by employing various parameters that can determine the sequence information. At present, the various methods are broadly used for prediction and representation of protein types. Pseudo amino acid (PseAA) composition and covariant discriminate algorithm were used by (Chou, 2001) in the prediction of membrane protein types. While, Fourier transform and support vector machine (SVM) by (Liu, et al., 2005) weighted SVM and PseAA composition by (Wang, et al., 2004). Discrete wavelets transform (DWT) and cascaded neural network by (Rezaei, et al., 2008) DWT and SVM by (Qiu, et al., 2010), while (wang, et al., 2010), in another work used dipeptide and back propagation network (BPN) for membrane protein type prediction. Various studies have been reported to identify the virulence factors through comparative genomics or homology searching approaches such as BLAST (Basic local alignment search tool ) (Altschul, et al., 1998) and by using machine learning approaches such as SPAAN (Software program for prediction of adhesins and adhesin-like proteins using neural networks) for adhesion protein identification (Sachdeva, et al., 2005) and VICMpred (Saha and Raghava, 2006) for bacterial virulent proteins classification. Recently, VirulentPred (Garg and Gupta, 2008) and Virulent- GO (Tsai, et al., 2009) used a bilayer cascade and gene-ontology in support vectors machine (SVM) classifier to predict the bacterial proteins that are involved in virulence.
However, these works are less focused to reveal the important features such as amino acid properties that plays a significant role as informative features to determine the protein virulence property (Chou and Cai, 2005; Chou and Shen, 2007). In this scenario, we focused to establish the advantages of combining the amino acid feature (composite model) and the ability of the two most distinguishing algorithms like SVM and BPN. Thus in this study, the amino acid composition (AAC), dipeptide composition (DPC) and combined version of the methods are employed as vectors to discriminate the features. While Support vector machine (SVM) and back propagation network (BPN) are base learners for classiïìÃÂcation and ten-fold cross-validation was applied to evaluate their performance.
Dataset construction
The bacterial virulent protein sequences were retrieved from VFDB (an integrated and comprehensive database of virulence factors of bacterial pathogens) (Chen, et al., 2005). The dataset was refined to filter the similar sequences by using PROSET (a fast procedure to create non-redundant sets of protein sequences) (Brendel, 1990). The final non-redundant dataset of 2051 comprises 1021 virulent sequences (positive dataset) and 1030 nonvirulent sequences (Negative dataset). This dataset was used to explore the classiïìÃÂcation capabilities of SVM and BPN. The overview of study is outlined in Figure. 1.

Figure 1: The flow chart of the work employed in this study.
Input features
Amino Acid Composition (AAC)
The compositions of amino acid representing a protein are considered as 20 dimensions feature vectors (Bhasin and Raghava, 2004). The amino acid composition is calculated by
AAC = Total Occurrence of ith amino acid in the sequence, Where i is single amino acid
Dipeptide Composition (DPC)
In this method, the occurrence of two adjacent amino acid residues that represents a protein by a vector of 400-dimension feature vectors (Chou, 1995). It takes an advantage over AAC of using sequence order information. The dipeptide composition of protein sequence is calculated as
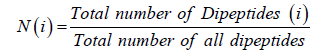
Where N (i) is singles dipeptide
Composite analysis
The combination of amino acid composition (AAC) and dipeptide Composition (DPC) features were used to train the both SVM and BPN classifiers. In this method, an input vector of 420 dimensions (AAC 20 features + 400 DPC features) was used to train the classifiers (Nakashima, et al., 1986).
Prediction algorithms
Support vector machine
The noise handling, large dataset and large feature space abilities of SVM has put forth its usage as a successful machine learning (ML)-technique in the field of bioinformatics and computational biology (Zavaljevski, et al., 2002). SVM classification can separate the positive points from negative points with higher margin. The parameters and kernels (linear, polynomial, radial base function (RBF) and sigmoid) were optimized for the best performance of SVM classifiers and trained with AAC and DPC features.
Back Propagation Network (BPN)
Back Propagation Network (BPN) uses gradient descent based delta learning rule (known as back propagation) for training the artificial neural networks (Russell and Norvig, 2003). This systematic method is computationally efficient in changing the weights in the network with function units to study a set of input-output patterns. This method can minimize the total squared error of the output. This trained supervised learning network can balance the ability to correctly respond to the input patterns (Figures. 1 and 2).

Figure 2: BPN model layout (architecture).
The determined amino acids contents of proteins are used as input patterns for training the Back Propagation Network. This BPN is a three layer network as shown in Figure. 2 corresponding to input, hidden and output layers (Basheer and Hajmeer, 2002). The input and output layer nodes are set to 20 and 1 respectively. The weight matrics (70 X 20) and bias matrix (1 X 20) connects input and hidden layers respectively. The four phases of training algorithms are weight initialization, feed forward, errors back propagation, weights and bias updation.
Cross validation and performance assessment
The jackknife cross-validation (Varma, et al., 2006) is established as one of the most effective and object oriented methods to evaluate a effectiveness of classifier in most of the statistical predictions. In this study, we employed ten-fold cross-validation to evaluate the SVM and BPM classifiers performance. We have divided the training dataset into 10 random subsets as that each subset consisting of equal number of virulent and non-virulent proteins. Then the nine sets were used to train the classifier while the performance of classifier was assessed on the one left subset. This was iterated ten times as subsets were included in training and test sets. Finally, the average performance was considered as the final performance of a classifier.
In general the performance of a prediction method is determined by threshold independent or threshold dependent parameters, while each has their own limitations. In this study, the threshold dependent parameters such as accuracy, sensitivity and specificity were measured to evaluate the prediction accuracy of each test dataset.
In this study, virulent proteins are defined as positive and non-virulent proteins as negative. To assess the sensitivity, specificity and accuracy true positives (TP), true negatives (TN), false positives (FP) and false negatives (FN) were determined.
Sensitivity (Sn): The classifiers ability in predicting the correct results are measured
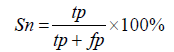
Specificity (Sp): The classifiers ability in predicting the incorrect results are measured.
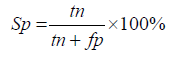
Accuracy (Acc): The classifiers ability to measure the degree of correctness of the predicted results to its actual value are measured
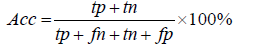
The availability of complete bacterial genomes of pathogens is a rich source of information to determine the virulence factors and its associated proteins. However, due to the complexity in determining the virulence factors, the urgency in computational tools to predict the virulence factors are desperately needed (Zheng, et al., 2012). In line with this several predicting machine learning algorithms have been proposed by many research groups to deal this prediction strategy. Thus in this study, the predictive capability of SVM and BPN algorithms and their reliability were determined by a widely used crossvalidation test that are widely used in the statistical methods. Also the comparative study of the feature representation methods are analyzed along with these classiïìÃÂcation methods and their performance are evaluated.
Prediction algorithms
The SVM classifier was trained and optimized with AAC, DPC and composite (AAC+DPC) features. Various kernels such as linear, quadratic, polynomial, radial basis function (RBF), MLP and RBF_sigma were optimized for the best performance of the SVM classifiers. The kernel parameters (C and gamma) corresponding to maximum accuracy were optimized as best parameter values. The detailed results of the kernel parameters for each AAC features are given in Table 1. The AAC-SVM classifier optimized with RBF kernel has the highest accuracy of 79.13% followed by Polynomial kernel with 78.54%. The lowest accuracy of 52.38 % was exhibited by MLP kernel. The average accuracy was considered for the best performance of a kernel. Thus in the present study, RBF kernel was considered as the most appropriate kernel in the SVM classifier -training and testing with AAC features (20 vectors).
| Kernels | Accuracy | Sensitivity | Fprate | Precision | Recall | Fmeasure | Specificity |
|---|---|---|---|---|---|---|---|
| Amino Acid Composition | |||||||
| Linear | 0.59 | 0.59 | 0.49 | 0.59 | 0.59 | 0.59 | 0.00 |
| Quadratic | 0.61 | 0.55 | 0.47 | 0.54 | 0.55 | 0.54 | 0.07 |
| Poly | 0.78 | 0.51 | 0.15 | 0.59 | 0.51 | 0.55 | 0.34 |
| RBF | 0.79 | 0.39 | 0.00 | 0.60 | 0.39 | 0.45 | 0.49 |
| MLP | 0.52 | 0.36 | 0.28 | 0.59 | 0.36 | 0.44 | 0.21 |
| RBF_Sigma | 0.59 | 0.59 | 0.49 | 0.59 | 0.59 | 0.59 | 0.00 |
Table 1. AAC-SVM classifier optimized with various kernels
In dipeptides composition method, the 400 features were used as input. The detailed results of DPC-SVM classifier optimized with various kernels were given in Table 2. It is observed that the highest accuracy of 88.07% is exhibited by Quadratic kernel, while the RBF kernel exhibited the lowest accuracy of 39.85 %. The average accuracy was considered for the best performance of a kernel. The high significant difference in the accuracy while using RBF kernels in the AAC and DPC classifiers may be owing to the low occurrence of possible dipeptides. Thus, the usage of quadratic kernel may be considered for the SVM classifier training and testing with DPC features (400 vectors).
| Kernels | Accuracy | Sensitivity | Fp rate | Precision | Recall | F measure | Specificity |
|---|---|---|---|---|---|---|---|
| Dipeptide Composition | |||||||
| Linear | 0.85 | 0.55 | 0.14 | 0.59 | 0.55 | 0.57 | 0.35 |
| Quadratic | 0.88 | 0.48 | 0.01 | 0.59 | 0.48 | 0.52 | 0.48 |
| Poly | 0.61 | 0.20 | 0.00 | 0.60 | 0.20 | 0.27 | 0.49 |
| RBF | 0.39 | 0.40 | 0.50 | 0.59 | 0.40 | 0.43 | 0.00 |
| MLP | 0.61 | 0.37 | 0.22 | 0.59 | 0.37 | 0.45 | 0.27 |
| RBF_Sigma | 0.85 | 0.55 | 0.14 | 0.59 | 0.55 | 0.57 | 0.35 |
Table 2. DPC-SVM classifier optimized with various kernels
Further, we made an attempt to enhance the prediction accuracy, the SVM classifier was trained with composite features as 420 vectors (AAC +DPC). The SVM classifier training and testing with composite features were also optimized with various kernel parameters (Table 3). It is observed that the RBF-Sigma kernel exhibited the highest accuracy of 89.27 % followed by quadratic kernel with 87.75 % of accuracy. The accuracy, sensitivity and specificity were shown in Figure. 3. Interestingly, the increase in 1.5 % accuracy was observed for composite SVM classifier with RBF-Sigma kernel when compared to that of AAC (RBF) and DPC (quadratic) SVM classifiers (Figure. 3). The average accuracy was considered for the best performance of a kernel. Thus the RBF-Sigma kernel was considered for the SVM classifier training and testing with composite features. This significantly suggests that the number of features and the kernel parameters pays a significant role in prediction performance of SVM algorithm. Thus suggesting RBF for AAC features; Quadratic for DPC features and RBF-Sigma for Composite features as best kernels.

Figure 3: The performance evaluation of SVM classifier for AAC, DPC and Composite method with various kernels.
| Kernels | Accuracy | Sensitivity | Fp rate | Precision | Recall | F measure | Specificity |
|---|---|---|---|---|---|---|---|
| AAC + DPC | |||||||
| Linear | 0.86 | 0.53 | 0.07 | 0.59 | 0.53 | 0.56 | 0.42 |
| Quadratic | 0.87 | 0.48 | 0.01 | 0.59 | 0.48 | 0.52 | 0.48 |
| Poly | 0.52 | 0.12 | 0.00 | 0.60 | 0.12 | 0.19 | 0.49 |
| RBF | 0.65 | 0.56 | 0.40 | 0.59 | 0.56 | 0.57 | 0.10 |
| MLP | 0.70 | 0.46 | 0.20 | 0.59 | 0.46 | 0.51 | 0.29 |
| RBF_Sigma | 0.89 | 0.53 | 0.07 | 0.59 | 0.53 | 0.56 | 0.42 |
Table 3. Composite-SVM classifier optimized with various kernels
Further, the effect of feature representation methods (AAC, DPC and Composite) on the classification ability of SVM and BPN were evaluated. The comparative results of SVM and BPN using AAC (RBF kernel), DPC (quadratic kernel) and composite (RBF-Sigma) model are shown in Table 4. Primarily, the classiïìÃÂers trained with AAC features revealed the accuracy of 79.13 % for SVM, while it is of 87% for BPN, which significantly evidenced that, the overall performance in terms of accuracy, sensitivity and specificity of BPN is higher than that of SVM. Thus suggesting the usage of BPN classifier would result in the accurate prediction of protein sequence while using their AAC features as input vectors.
| Methods | Accuracy | Sensitivity | Fp rate | Precision | Recall | F measure | Specificity |
|---|---|---|---|---|---|---|---|
| AAC | |||||||
| SVM (RBF) | 0.79 | 0.39 | 0.00 | 0.60 | 0.39 | 0.45 | 0.49 |
| BPN | 0.87 | 0.55 | 0.09 | 0.60 | 0.55 | 0.57 | 0.41 |
| DPC | |||||||
| SVM (Quadratic) | 0.88 | 0.48 | 0.01 | 0.59 | 0.48 | 0.52 | 0.48 |
| BPN | 0.87 | 0.51 | 0.09 | 0.59 | 0.51 | 0.54 | 0.41 |
| Composite | |||||||
| SVM (RBF_sigma) | 0.89 | 0.53 | 0.07 | 0.59 | 0.53 | 0.56 | 0.42 |
| BPN | 0.86 | 0.52 | 0.10 | 0.59 | 0.52 | 0.55 | 0.40 |
Table 4. Comparative performances of SVM and BPN using AAC, DPC and composite model classifier optimized under the best kernels respectively
It is observed that the total amino acid Composition analysis revealed the presence of Leucine, Alanine, Glycine, Serine and Valine as in most frequently occurring amino acids in the sequence. Interestingly the high number of Leucine is observed in both virulent and non-virulent sequences. The high composition of amino acid such as Alanine (A), Cystine (C), Glutamic acid (E), Phenyl alanine (F), Glycine (G), Histdine (H), Isoleucine (I), Leucine (L), Methionine (M), Proline (P), Arginine (R), Valine (V) and Tryptophan (W) were observed in the protein sequences that are classified as non-virulent proteins, while the remaining amino acids such as Aspargine (N), Glutamine (Q), Serine (S), Threonine (T) and Tyrosine (Y) were high in the protein sequences predicted as virulent which significantly implies that these residual compositions plays a major role in the successful classification of models that can predict the sequence as virulent and non-virulent.
In another discrete method, the classifiers are trained on dipeptide composition (DPC) that is used to represent the protein sequence. However, DPC has shown a greater improvement in the performance of SVM classier with the highest accuracy of 88.07% which is relatively higher than the accuracy of BPN classifier. It means SVM classifiers is reliably good while using quadratic kernel in discriminating the proteins based on their dipeptide composition, as these dipeptides provides the information regarding amino acid composition as well as their local order. However, the low sensitivity and specificity value of SVM suggests that it might be due to the low frequency occurrence of dipeptides in the dataset.
While the performance in terms of sensitivity and specificity of BPN is higher than that of SVM, significantly suggests its usage as best classifier in predicting the proteins while using dipeptide composition. While using the dipeptide composition analysis as feature, the presence of dipeptides such as AC, AL, VL, LL, SG, LV, TL is observed as most frequently occurring dipeptides in the virulent sequence, while LL, LA, AL, AA, VL, VA, LV, LG, GL, and AG in non-virulent proteins.
In order to enhance the performance of classiïìÃÂers further, the composite model of AAC and DPC were proposed in this study. A notable performance has been observed in the classiïìÃÂers while using this proposed composite model. It is observed that SVM has yielded higher success rate with 89.27 % while compared to that of BPN with the prediction accuracy of 86 %. However, the overall performance of SVM in terms of accuracy, sensitivity and specificity is found to be higher than that of BPN performance (Figure. 4). Many previous reports have significantly stated that the prediction methods based on compositional features are more accurate in predicting the function, secondary structures and sub-cellular localization of proteins (Garg, et al., 2005; Kaur and Raghava, 2003). Similarly, in this study also it is observed that the AAC, DPC and the composite modules used in both SVM and BPN were found to have higher accuracy.

Figure 4: Overall performance of SVM and BPN methods.
The prediction accuracy of SVM is almost 10% and 3% higher than the BPN while using DPC and composite features. Whereas, the sensitivity and specificity of BPN exhibited huge difference SVM for all the AAC, DPC and composite methods. This significantly suggests that the even though the raise in performance of SVM classiïìÃÂers for DPC and composite model are observed the low sensitivity and specificity values may be due the discrimination power of the large number of feature extraction strategies. Thus suggesting the usages of BPN over SVM classifiers as the best classifier for predicting the proteins sequence based on their compositions (Figure. 4).
The discriminate power of the classifiers evaluated by various statistical parameters is appropriate for both the methods while considering the kernels, which significantly implies that there is no statistically significant difference in the accuracy in the evaluated methods, which further envisages that the usage of BPN method over SVM to be the better choice of prediction algorithm to predict the protein sequence as virulent. Taking in to the account of total accuracy, sensitivity and specificity of SVM and BPN classifiers the unbalance of classification efficiency in predicting the virulence and non-virulence of protein sequences are observed. It is also observed that while using SVM classifiers, the accuracy varies from features to feature while using kernel parameter suggesting that few kernels are good in predicting the sequences with high sensitivity and some kernels with the highest specificity values indicating their good ability in predicting the sequences. However, the performance of exhibited by the BPN classifiers makes it as a choice of best classifying algorithm. In line with this the oldest fishers linear discriminate analysis that ranks much greater than over each method indicates the performance limits of various kernels and classifiers assessed in this study. Also suggesting that the optimization of parameters will result in high discriminating power of SVM and BPN classifiers.
It is noteworthy to mention that the results obtained in this study are based on a specific data set. It is well known that the performance of SVM classifier and its prediction ability are the dependents of their various kernel parameters. Although various kernels are used to optimize the SVM classifier, to minimize the total error rates it is clearly observed that the performance of the classifier is just a reflection of the chosen parameters. It is worth to mention that the limitations of this study may be the bigger dataset that could eventually lead to different and improved results respectively. Thus this study provides a comprehensive analytic frame work of employing the better classifier and also the potential features that can significantly implemented as ML algorithms in prediction.
Comparison with other methods
To the best of our knowledge, there is no report on the comparison of ML algorithms that classify protein sequences into virulent and non-virulent. However, Chou reported the usage of pseudo amino acid (PseAA) composition and covariant discriminate algorithm in the prediction of membrane protein types. While SPAAN, VICMpred, VirulentPred and Virulent-GO are the machine learning approaches that are used for bacterial virulent proteins classification. These systems used single and cascade feature in support vectors machine (SVM) classifier for their predictions. In contrast, the proposed approach has showed the significant increase in accuracy while using RBF, Quadratic and RBF-Sigma kernels for AAC, DPC and composite methods respectively. Further, the results show that the performance of BPN approach is signiïìÃÂcantly higher than SVM while using AAC, DPC and composite methods in classifying protein sequence as virulent and non-virulent.
Predicting the protein sequence as virulent factor plays a key role in the development of novel drug to combat with many dreadful diseases. In this study, various kernel parameters of SVM classifier were evaluated on the non-redundant dataset protein sequences. The amino acid composition, dipeptide composition and composite methods were used as input features. Ten-fold cross validation was applied to evaluate the performance of SVM and BPN classifiers and measured with standard parameters like accuracy, sensitivity, false positive rate, precision, recall, f-measure and specificity. It is observed that the BPN classifier exhibited higher accuracy with AAC features, while SVM exhibited higher accuracy over BPN by using DPC and composite methods. But taking into the account of sensitivity and specificity, the BPN classifier exhibited better performance over SVM while using DPC and composite methods, which are higher than that of sensitivity and specificity values of SVM classifier. Thus this study provides information on general tendency of protein sequence dataset and suggests the researchers to select the best classifier and its optimization. Also it provides insights into future researcher to avoid assessment of data by using only one method and also suggests choosing the optimal ML algorithms for virulent researchers.
Copyright © 2026 Research and Reviews, All Rights Reserved